About#
I started working on this page in 2018. In 2022 I rewrote the page and removed the old posts (some were starting to be somewhat outdated). Originally, my main motivations for creating this page were that I wanted to:
keep track of my work and how different “parts” connect,
keep track of my progress when learning new things (basically, I wanted to write short notes to remind myself of things I learn which I think will be useful later),
write short notes about other things, again mainly for me, but they could maybe be of interest to others,
learn more about how to make & set up a webpage.
For keeping track of my work, I have made a timeline (see below) showing the places I have worked and what I have done. There is also an overview of articles I have co-authored.
Where I have worked#
2019 – present
Associate Professor, Department of Chemistry, NTNU, Trondheim.
Keywords: Molecular Dynamics | Monte Carlos | Non-equilibrium thermodynamics | Rare events | Biological systems
I am currently working as an associate professor at the Department of Chemistry, NTNU. My main topics are presently directed towards non-equilibrium thermodynamics, biological systems, understanding reactions and performing simulations for understanding natural phenomena.

2019 – 2019
Researcher, Norwegian Defence Research Establishment, FFI, Lillestrøm.
Keywords: Modeling | Simulations
I worked at FFI on modeling and simulations, and visualization of simulations results. Part of my work was directed towards understanding ballistic threats.

2016 – 2019
Researcher, Department of Chemistry, NTNU, Trondheim.
Keywords: Molecular Dynamics | Monte Carlo | Rare events | Protein simulation
I'm worked as a researcher in the Theoretical Chemistry section, collaborating with colleagues at the department of biotechnology. We studied how a specific protein (illustrated in the image) converts between different alginate polymers. Alginate (which is the equivalent of cellulose for seaweed) has many applications within pharmaceuticals, food, and cosmetics. The proteins we studied can make specific alginate polymers and the specificity can be modified by mutating different amino acids. My work included computer simulations to understand the operation of the protein and how different mutations influence it.

2014 – 2016
Postdoctoral Fellow, Department of Chemistry, NTNU, Trondheim.
Keywords: Molecular Dynamics | Monte Carlo | Rare events | Transition path sampling | Code development
I was part of the Theoretical Chemistry section, working in the group of Professor Titus van Erp. We developed new computational methods with the goal of expanding the accessible time- and system scales for computer simulations of complex systems. In simpler terms, for me at least, this means that we worked on methods that can be used to efficiently (and correctly) study reactions. One outcome of our work is the PyRETIS Python package for simulations of rare events.
2012 – 2014
Research Associate, Department of Chemistry, Imperial College London.
Keywords: Molecular Dynamics | Non-equilibrium thermodynamics | Transport phenomena | Phase transitions | Molecular machines
I was part of the Computational Chemical Physics group, working with Professor Fernando Bresme from October 2012 to March 2014. The main focus of our work was non-equilibrium transport phenomena, specifically coupling between mass, heat and charge transport. The image shows water molecules under a temperature gradient. Under these conditions, the molecules will orient and give rise to an electric field. As a part of my work, we studied the conditions for this orientation.

2008 – 2012
Ph.D., Department of Chemistry, NTNU.
Keywords: Molecular Dynamics | Non-equilibrium thermodynamics | Molecular machines | Protein simulation | Energy dissipation
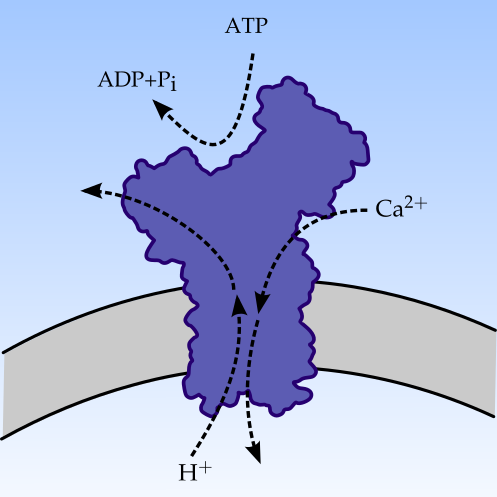
My thesis is titled Energy dissipation in biomolecular machines and was supervised by Professor Signe Kjelstrup. My Ph.D. project was funded by the Faculty of Natural Sciences and Technology, NTNU, and included teaching duties equivalent to a full year of teaching. The protein in the image is the calcium pump which we studied in detail. We developed both theoretical models for describing the operation of the pump and MD methodologies for investigating heat transfer and dissipation in such nano machines.
List of articles#
Below is a list of articles, sorted on years, with a short summary.
2022 (3)#
Computational study of the dissolution of cellulose into single chains: the role of the solvent and agitation
Eivind Bering, Jonathan Ø. Torstensen, Anders Lervik and Astrid S. de Wijn
Cellulose, 29:1365-1380, 2022 | Link to article
View summary
We investigate the dissolution mechanism of cellulose using molecular dynamics simulations in both water and a mixture solvent consisting of water with Na⁺, OH⁻ and urea. As a first computational study of its kind, we apply periodic external forces that mimic agitation of the suspension. Without the agitation, the bundles do not dissolve, neither in water nor solvent. In the solvent mixture the bundle swells with significant amounts of urea entering the bundle, as well as more water than in the bundles subjected to pure water. We also find that the mixture solution stabilizes cellulose sheets, while in water these immediately collapse into bundles. Under agitation the bundles dissolve more easily in the solvent mixture than in water, where sheets of cellulose remain that are bound together through hydrophobic interactions. Our findings highlight the importance of urea in the solvent, as well as the hydrophobic interactions, and are consistent with experimental results.
Phonon thermal transport in copper: The effect of size, crystal orientation, and grain boundaries
Sandra Sæther, Merete Falck Erichsen, Senbo Xiao, Zhiliang Zhang, Anders Lervik and Jianying He
AIP Advances, 12:065301, 2022 | Link to article
View summary
In electronic devices at the micro- and nanoscale, thermal management is vital. At such small sizes, crystal orientation, grain boundaries, and even the size itself can play an important role in the thermal transport and need to be taken into careful consideration when devices are designed. In this article, we perform computational experiments using non-equilibrium molecular dynamics simulations to evaluate the effect of size, orientation, and grain boundaries on the phonon thermal transport of copper. In addition, we compare the results obtained from the rescale and Langevin thermostat procedures. We find that the contribution of phonons to the total thermal conductivity in copper increases as the size decreases. Furthermore, the Σ5(210)[001] twist grain boundary is found to have a significant effect on the thermal transport of a bi-crystalline copper system when the grains are 15 nm. No such effect is found for the Σ3(112)[110] twin boundary. The effect of crystal orientation on the thermal conductivity is also studied, and no discerned effect can be observed. It is found that the Langevin thermostat leads to an over-estimation of the thermal conductivities at smaller scales and should be used with caution.
The role of pressure and defects in the wurtzite to rock salt transition in cadmium selenide
Anders Lervik, Ingeborg-Helene Svenum, Zhaohui Wang, Raffaela Cabriolu, Enrico Riccardi, Stefan Andersson and Titus S. van Erp
Phys. Chem. Chem. Phys., 24:8378-8386, 2022 | Link to article
View summary
Using molecular dynamics and path sampling techniques we investigated the effect of pressure and defects in the wurtzite to rock salt transition in cadmium selenide (CdSe). In the pressure range 2–10 GPa, rate constants of transition are in the order of 10−23 to 105 s−1 for the transformation of a relatively small wurtzite crystal consisting of 1024 atoms with periodic boundary conditions. The transition paths predominantly evolve through an intermediate 5-coordinated structure, as reported before, though its typical lifetime within the transition paths is particularly long in the intermediate pressure range (4–6 GPa). The defects were created by removing Cd–Se pairs from an otherwise perfect crystal. The removals were either selected fully randomized or grouped in clusters (cavity creation). We find that the rate of transition due to the defects increases by several orders of magnitude even for a single pair removal. This is caused by a change in the transition mechanism that no longer proceeds via the intermediate 5-coordinated structure, when defects are present. Further, the cavity creation yields a lower rate than the fully randomized removal.
2021 (3)#
Thermal Transport in Polyethylene: The Effect of Force Fields and Crystallinity
Sandra Sæther, Merete Falck, Zhiliang Zhang, Anders Lervik and Jianying He
Macromolecules, 54:6563-6574, 2021 | Link to article
View summary
In this article, we study the local structure and heat transfer properties (thermal conductivity and interfacial conductance) in model semi-crystalline polyethylene (PE) by non-equilibrium molecular dynamics. We compare three different force fields with different levels of detail (all-atom, all-atom with constraints, and united-atom) and find that the structure of the model PE is significantly influenced by the choice of force field. The united-atom force field results in a reduced overall crystallinity and an over-idealized organization of the polymer chains, compared to the all-atom force fields. We find that thermal transport properties are not greatly influenced when structural effects are taken into consideration, and our results suggest that united-atom models can be used to study heat transfer properties of model PE, with decreased computational cost.
Alginate gels crosslinked with chitosan oligomers – a systematic investigation into alginate block structure and chitosan oligomer interaction
Georg Kopplin, Anders Lervik, Kurt I. Draget and Finn L. Aachmann
RSC Adv., 11:13780-13798, 2021 | Link to article
View summary
Three alginates with fundamentally different block structures, poly-M, poly-G, and poly-MG, have been investigated upon ionic crosslinking with chitosan oligosaccharides (CHOS), using circular dichroism (CD), rheology, and computer simulations, supporting the previously proposed gelling principle of poly-M forming zipper-like junction zones with chitosan (match in charge distance along the two polyelectrolytes) and revealing a unique high gel strength poly-MG chitosan gelling system. CD spectroscopy revealed an increased chiroptical activity exclusively for the poly-M chitosan gelling system, indicative of induced conformational changes and higher ordered structures. Rheological measurement revealed gel strengths (G′ < 900 Pa) for poly-MG (1%) CHOS (0.3%) hydrogels, magnitudes of order greater than displayed by its poly-M analogue. Furthermore, the ionically crosslinked poly-MG chitosan hydrogel increased in gel strength upon the addition of salt (G′ < 1600 at 50 mM NaCl), suggesting a stabilization of the junction zones through hydrophobic interactions and/or a phase separation. Molecular dynamics simulations have been used to further investigate these findings, comparing interaction energies, charge distances and chain alignments. These alginates are displaying high gel strengths, are known to be fully biocompatible and have revealed a broad range of tolerance to salt concentrations present in biological systems, proving high relevance for biomedical applications.
The energy conversion in active transport of ions
Signe Kjelstrup and Anders Lervik
Proceedings of the National Academy of Sciences, 118:e2116586118, 2021 | Link to article
View summary
In this article, we comment on the energy conversion in active transport and the recent simulation results of Kobayashi et al.
2020 (3)#
PyRETIS 2: An improbability drive for rare events
Enrico Riccardi, Anders Lervik, Sander Roet, Ola Aarøen and Titus S. van Erp
J. Comput. Chem., 41:370-377, 2020 | Link to article
View summary
The algorithmic development in the field of path sampling has made tremendous progress in recent years. Although the original transition path sampling method was mostly used as a qualitative tool to sample reaction paths, the more recent family of interface-based path sampling methods has paved the way for more quantitative rate calculation studies. Of the exact methods, the replica exchange transition interface sampling (RETIS) method is the most efficient, but rather difficult to implement. This has been the main motivation to develop the open-source Python-based computer library PyRETIS that was released in 2017. PyRETIS is designed to be easily interfaced with any molecular dynamics (MD) package using either classical or ab initio MD. In this study, we report on the principles and the software enhancements that are now included in PyRETIS 2, as well as the recent developments on the user interface, improvements of the efficiency via the implementation of new shooting moves, easier initialization procedures, analysis methods, and supported interfaced software. © 2019 The Authors. Journal of Computational Chemistry published by Wiley Periodicals, Inc.
Teaching complex molecular simulation algorithms: Using self-evaluation to tailor web-based exercises at an individual level
Oda Dahlen, Anders Lervik, Ola Aarøen, Raffaela Cabriolu, Reidar Lyng and Titus S. van Erp
Computer Applications in Engineering Education, 28:779-791, 2020 | Link to article
View summary
Abstract It is quite challenging to learn complex mathematical algorithms used in molecular simulations, stressing the importance of using the most advantageous teaching methods. Ideally, individuals should learn at their pace and deal with tasks fitting their levels. Web-based exercises make it possible to tailor every small step of the learning process, but this requires continuous monitoring of the learner. Differentiation based on the scores after the first round of common tasks can be demotivating for all students, as they will experience the initial set of tasks as being either too easy or too hard. We designed two tests, a self-monitoring test and a rapid test (RT) in which the students explained equations relating to the current topic. The first test was aimed to see if the students were able to evaluate their own level of knowledge, whereas the RT was aimed to find a fast way to determine the level of the students. We compared both tests with traditional measures of knowledge and used a relatively new method, which was originally designed for the analysis of molecular simulation data, to interpret the results. Based on this analysis, we concluded that self-evaluation, rather than an RT, is a valuable tool to automatically steer individual students through a tree of web-based exercises to match their skill levels and interests.
Identification of a Pivotal Residue for Determining the Block Structure-Forming Properties of Alginate C-5 Epimerases
Annalucia Stanisci, Anne Tøndervik, Margrethe Gaardløs, Anders Lervik, Gudmund Skjåk-Bræk, Håvard Sletta and Finn L. Aachmann
ACS Omega, 5:4352-4361, 2020 | Link to article
View summary
Alginate is a linear copolymer composed of 1→4 linked β-d-mannuronic acid (M) and its epimer α-l-guluronic acid (G). The polysaccharide is first produced as homopolymeric mannuronan and subsequently, at the polymer level, C-5 epimerases convert M residues to G residues. The bacterium Azotobacter vinelandii encodes a family of seven secreted and calcium ion-dependent mannuronan C-5 epimerases (AlgE1–AlgE7). These epimerases consist of two types of structural modules: the A-modules, which contain the catalytic site, and the R-modules, which influence activity through substrate and calcium binding. In this study, we rationally designed new hybrid mannuronan C-5 epimerases constituting the A-module from AlgE6 and the R-module from AlgE4. This led to a better understanding of the molecular mechanism determining differences in MG- and GG-block-forming properties of the enzymes. A long loop with either tyrosine or phenylalanine extruding from the β-helix of the enzyme proved essential in defining the final alginate block structure, probably by affecting substrate binding. Normal mode analysis of the A-module from AlgE6 supports the results.
2019 (2)#
Non-equilibrium thermodynamics as a tool to compute temperature at the catalyst surface
Carolina Cruz, Daniel Barragán, Elisa Magnanelli, Anders Lervik and Signe Kjelstrup
Phys. Chem. Chem. Phys., 21:15195-15205, 2019 | Link to article
View summary
The surface temperature is computed for a heterogeneous catalytic reaction model, namely the oxidation of carbon monoxide on platinum. The surface temperature was found using non-equilibrium thermodynamic theory, a theory which provides the proper dependencies between heat and mass fluxes and the reaction rate. The theory predicts a possible coupling between the reaction rate and the thermal driving force and can help extend classical reaction kinetics. In the absence of direct measurements, we explore the coupling numerically. The results are able to capture experimental data reported in the literature, and give new insights into why Arrhenius plots may turn out to be non-linear.
Molecular Structure and Solubility Determination of Asphaltenes
Salim Ok, Mehdi Mahmoodinia, Navvamani Rajasekaran, Mohamed A. Sabti, Anders Lervik, Titus S. van Erp and Raffaela Cabriolu
Energy & Fuels, 33:8259-8270, 2019 | Link to article
View summary
Due to their complex chemical structure, identification and characterization of asphaltenes remain a big challenge for researchers in the oil industry. In the current contribution, solution-state two-dimensional (2D) nuclear magnetic resonance (NMR) spectra of the asphaltenes, obtained from either heavy or light crude oil samples, provide information on the chemical structures of asphaltenes at the molecular level. Two-dimensional 1H–13C heteronuclear NMR measurements are particularly useful for differentiating among several structural environments whose signals overlap in the one-dimensional 13C spectra. Both, 2D 1H–13C heteronuclear and 1H–1H homonuclear NMR spectra indicate short- and long-range interactions between different functional groups of the asphaltene samples, revealing even “bay”- and “fjord”-type structures. Based on the NMR correlations between different protons and carbons with different chemical environments, we propose various chemical structures (polyaromatic cores with aliphatic chains, porphyrin derivatives, organic salts). Furthermore, through molecular dynamics simulations, we have obtained Hildebrand solubility parameters of different asphaltene solvents and of two experimental recovered asphaltene structures. Our calculated solubilities are consistent with previous experimental and simulation works.
2018 (2)#
Local initiation conditions for water autoionization
Mahmoud Moqadam, Anders Lervik, Enrico Riccardi, Vishwesh Venkatraman, Bjørn K ̊are Alsberg and Titus S. van Erp
Proceedings of the National Academy of Sciences, 2018 | Link to article
View summary
The dissociation of water is arguably the most fundamental chemical reaction occurring in the aqueous phase. Despite that the splitting of a water molecule very seldom occurs, the reaction is of major importance in many areas of chemistry and biology. Direct experimental probing of the event is still impossible and also simulating the event via accurate computer simulations is challenging. Here, we achieved the latter via specialized rare-event algorithms estimating rates of dissociation in agreement with indirect experimental measurements. Even more interestingly, by a rigorous analysis of our results we identified anomalies in the water structure that act as initiators of the reaction, a finding that suggests paradigms for steering and catalyzing chemical reactions.The pH of liquid water is determined by the infrequent process in which water molecules split into short-lived hydroxide and hydronium ions. This reaction is difficult to probe experimentally and challenging to simulate. One of the open questions is whether the local water structure around a slightly stretched OH bond is actually initiating the eventual breakage of this bond or whether this event is driven by a global ordering that involves many water molecules far away from the reaction center. Here, we investigated the self-ionization of water at room temperature by rare-event ab initio molecular dynamics and obtained autoionization rates and activation energies in good agreement with experiments. Based on the analysis of thousands of molecular trajectories, we identified a couple of local order parameters and show that if a bond stretch occurs when all these parameters are around their ideal range, the chance for the first dissociation step (double-proton jump) increases from 10-7 to 0.4. Understanding these initiation triggers might ultimately allow the steering of chemical reactions.
Temperature anisotropy at equilibrium reveals nonlocal entropic contributions to interfacial properties
Øivind Wilhelmsen, Thuat T. Trinh and Anders Lervik
Phys. Rev. E, 97:012126, 2018 | Link to article
View summary
Density gradient theory for fluids has played a key role in the study of interfacial phenomena for a century. In this work, we revisit its fundamentals by examining the vapor-liquid interface of argon, represented by the cut and shifted Lennard-Jones fluid. The starting point has traditionally been a Helmholtz energy functional using mass densities as arguments. By using rather the internal energy as starting point and including the entropy density as an additional argument, following thereby the phenomenological approach from classical thermodynamics, the extended theory suggests that the configurational part of the temperature has different contributions from the parallel and perpendicular directions at the interface, even at equilibrium. We find a similar anisotropy by examining the configurational temperature in molecular dynamics simulations and obtain a qualitative agreement between theory and simulations. The extended theory shows that the temperature anisotropy originates in nonlocal entropic contributions, which are currently missing from the classical theory. The nonlocal entropic contributions discussed in this work are likely to play a role in the description of both equilibrium and nonequilibrium properties of interfaces. At equilibrium, they influence the temperature- and curvature-dependence of the surface tension. Across the vapor-liquid interface of the Lennard Jones fluid, we find that the maximum in the temperature anisotropy coincides precisely with the maximum in the thermal resistivity relative to the equimolar surface, where the integral of the thermal resistivity gives the Kapitza resistance. This links the temperature anisotropy at equilibrium to the Kapitza resistance of the vapor-liquid interface at nonequilibrium.
2017 (3)#
Entropy facilitated active transport
J. Miguel Rubí, Anders Lervik, Dick Bedeaux and Signe Kjelstrup
J. Chem. Phys., 146:185101, 2017 | Link to article
View summary
We show how active transport of ions can be interpreted as an entropy facilitated process. In this interpretation, a particular change in the pore geometry through which substrates are transported gives rise to a driving force. This chemical energy provided by the chemical reaction is then used to create a protein geometry favorable for the uphill transport of ions. Attempts to estimate the energy available by this change in several proteins shows that an entropic contribution from the pore geometry is significant. We discuss how this effect can be used to understand how energy transduction in active transport can take place over a relatively long distance.
PyRETIS: A well-done, medium-sized python library for rare events
Anders Lervik, Enrico Riccardi and Titus S. van Erp
J. Comput. Chem., 38:2439--2451, 2017 | Link to article
View summary
Transition path sampling techniques are becoming common approaches in the study of rare events at the molecular scale. More efficient methods, such as transition interface sampling (TIS) and replica exchange transition interface sampling (RETIS), allow the investigation of rare events, for example, chemical reactions and structural/morphological transitions, in a reasonable computational time. Here, we present PyRETIS, a Python library for performing TIS and RETIS simulations. PyRETIS directs molecular dynamics (MD) simulations in order to sample rare events with unbiased dynamics. PyRETIS is designed to be easily interfaced with any molecular simulation package and in the present release, it has been interfaced with GROMACS and CP2K, for classical and ab initio MD simulations, respectively.
Rare event simulations reveal subtle key steps in aqueous silicate condensation
Mahmoud Moqadam, Enrico Riccardi, Thuat T. Trinh, Anders Lervik and Titus S. van Erp
Phys. Chem. Chem. Phys., 19:13361-13371, 2017 | Link to article
View summary
A replica exchange transition interface sampling (RETIS) study combined with Born-Oppenheimer molecular dynamics (BOMD) is used to investigate the dynamics, thermodynamics and the mechanism of the early stages of the silicate condensation process. In this process, two silicate monomers, of which one is an anionic species, form a negatively charged five-coordinated silicate dimer. In a second stage, this dimer can fall apart again, forming the original monomers, or release a water molecule into the solution. We studied the association and dissociation reaction in the gas phase, and the dissociation and water removal step in the aqueous phase. The results on the aqueous phase dissociation suggest two possible mechanisms. The breakage of the bond between the intermediate oxygen and the five-coordinated silicon is sometimes accompanied by a proton transfer. After dissociation into silicate monomers, the anionic monomer is either the previously four-coordinated silicon or the previously five-coordinated silicon depending on whether the hydrogen transfer occurs or not. Our results show that the mechanism of proton transfer is highly predominant. Water removal simulations also show two possible mechanisms distinguished by the proton transfer reaction path. Proton transfer can occur either via a direct or via a water mediated reaction step. The calculations reveal that although both mechanisms contribute to the water removal process, the direct proton transfer is slightly favorable and occurs roughly in six out of ten occasions.
2016 (6)#
Coherent description of transport across the water interface: From nanodroplets to climate models
Øivind Wilhelmsen, Thuat T. Trinh, Anders Lervik, Vijay Kumar Badam, Signe Kjelstrup and Dick Bedeaux
Phys. Rev. E, 93:032801, 2016 | Link to article
View summary
Transport of mass and energy across the vapor-liquid interface of water is of central importance in a variety of contexts such as climate models, weather forecasts, and power plants. We provide a complete description of the transport properties of the vapor-liquid interface of water with the framework of nonequilibrium thermodynamics. Transport across the planar interface is then described by 3 interface transfer coefficients where 9 more coefficients extend the description to curved interfaces. We obtain all coefficients in the range 260–560 K by taking advantage of water evaporation experiments at low temperatures, nonequilibrium molecular dynamics with the TIP4P/2005 rigid-water-molecule model at high temperatures, and square gradient theory to represent the whole range. Square gradient theory is used to link the region where experiments are possible (low vapor pressures) to the region where nonequilibrium molecular dynamics can be done (high vapor pressures). This enables a description of transport across the planar water interface, interfaces of bubbles, and droplets, as well as interfaces of water structures with complex geometries. The results are likely to improve the description of evaporation and condensation of water at widely different scales; they open a route to improve the understanding of nanodroplets on a small scale and the precision of climate models on a large scale.
Chapter 16 Mesoscopic Non-equilibrium Thermodynamics in Biology
Anders Lervik and J. Miguel Rubi
in Experimental Thermodynamics Volume X: Non-equilibrium Thermodynamics with Applications, pages 338-355. The Royal Society of Chemistry, 2016 | Link to article
View summary
This chapter deals with applications of mesoscopic non-equilibrium thermodynamics to biological systems, in particular to the study of kinetic processes and energy transduction mechanisms that take place in biological systems. The inherent non-linear nature of these processes makes it necessary to put the non-equilibrium thermodynamics method into a broader scope. We will show that by going to the mesoscale domain, the limitation of only providing linear laws can be removed and non-linear kinetic laws, characteristic of biochemical processes, can be obtained. We exemplify the case of enzyme catalysis through the Michaelis-Menten mechanism, and we consider energy transduction in proteins and the stretching of DNA molecules in detail. Mesososcopic non-equilibrium thermodynamics provides a general framework for the study of small-scale biological systems that operate far from equilibrium.
Analyzing Complex Reaction Mechanisms Using Path Sampling
Titus S. van Erp, Mahmoud Moqadam, Enrico Riccardi and Anders Lervik
J. Chem. Theory Comput., 12:5398-5410, 2016 | Link to article
View summary
We introduce an approach to analyze collective variables (CVs) regarding their predictive power for a reaction. The method is based on already available path sampling data produced by, for instance, transition interface sampling or forward flux sampling, which are path sampling methods used for efficient computation of reaction rates. By a search in CV space, a measure of predictiveness can be optimized and, in addition, the number of CVs can be reduced using projection operations which keep this measure invariant. The approach allows testing hypotheses on the reaction mechanism but could, in principle, also be used to construct the phase-space committor surfaces without the need of additional trajectory sampling. The procedure is illustrated for a one-dimensional double-well potential, a theoretical model for an ion-transfer reaction in which the solvent structure can lower the barrier, and an ab initio molecular dynamics study of water auto-ionization. The analysis technique enhances the quantitative interpretation of path sampling data which can provide clues on how chemical reactions can be steered in desired directions.
Chapter 6 Non-equilibrium Molecular Dynamics
Fernando Bresme, Anders Lervik and Jeff Armstrong
in Experimental Thermodynamics Volume X: Non-equilibrium Thermodynamics with Applications, pages 105-133. The Royal Society of Chemistry, 2016 | Link to article
View summary
This chapter discusses non-equilibrium molecular dynamics computer simulations. The focus is on the computation of coefficients that quantify transport properties (diffusion, thermal conductivity and viscosity) of simple and complex fluids as well as their interfaces. Several non-equilibrium methods are discussed, highlighting their connection to non-equilibrium thermodynamics. Coupling phenomena are also considered and applications to bulk fluids and interfaces are reviewed.
Note: A new truncation correction for the configurational temperature extends its applicability to interaction potentials with a discontinuous force
Anders Lervik, Øivind Wilhelmsen, Thuat T. Trinh and Edgar M. Blokhuis
J. Chem. Phys., 144:056101, 2016 | Link to article
View summary
We present a simple truncation correction for the configurational temperature which, unlike previous corrections, works even at low truncation values for the shifted and truncated Lennard-Jones potential. The success of the new correction suggests that the expression for the configurational temperature is valid also for interaction potentials with a discontinuous force, given that the discontinuity is properly accounted for.
Structural organization of sterol molecules in DPPC bilayers: a coarse-grained molecular dynamics investigation
Yawen Zhang, James W. Carter, Anders Lervik, Nicholas J. Brooks, John M. Seddon and Fernando Bresme
Soft Matter, 12:2108-2117, 2016 | Link to article
View summary
We investigate the structural organization of cholesterol (CHOL) analogues in 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) bilayers using coarse-grained molecular dynamics simulations and the MARTINI forcefield. Different sterol molecules are modelled by increasing (CHOLL) or decreasing (CHOLS) the diameter of the sterol beads employed in the MARTINI model of CHOL. At high sterol concentrations, (xsterol = 0.5), typical of liquid ordered phases, we find that the sterol arrangement and sterol-DPPC interactions strongly depend on the sterol size. Smaller sterols (CHOLS and CHOL) form linear clusters, while the larger sterols (CHOLL) arrange themselves into disc shaped clusters. By combining structural and dynamical properties we also investigate the So [rightward arrow] Ld transition for the CHOLL and CHOLS sterols. We show that small changes in the sterol size significantly affect the stability of the gel phase with the gel phase stabilized by the small sterols, but destabilized by large sterols. The general dependence of the phase behaviour of the membrane with sterol content is reminiscent of the one observed in naturally occurring membranes. The relevance of our results to understand current cholesterol-bilayer structural models is discussed.
2015 (4)#
A coarse-grained molecular dynamics investigation of the phase behavior of DPPC/cholesterol mixtures
Yawen Zhang, Anders Lervik, John Seddon and Fernando Bresme
Chem. Phys. Lipids, 185:88 - 98, 2015 | Link to article
View summary
We report a systematic molecular dynamics computer simulation investigation of the phase behavior of 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC)/cholesterol (CHOL) bilayers for a wide range of temperatures and cholesterol compositions (0–50%). Our simulations consist of several thousand phospholipids modeled with the coarse grained MARTINI forcefield covering timescales of tens of microseconds. We employ Voronoi and Delaunay constructions to quantify local compositions in an attempt to identify coexistence regions with the liquid ordered (Lo) phase. We combine this analysis with the chain order parameter and in plane pair correlation functions and propose a phase coexistence diagram for the DPPC/CHOL mixture. We find stable liquid ordered phases Lo for high cholesterol content (40–50%). At these concentrations the cholesterol molecules arrange themselves in linear clusters. Our simulations provide evidence for coexistence between the Lo and gel phases at low temperatures, while at high temperatures the transition from the liquid disordered phase to the Lo phase is continuous and coexistence is not observed. The lipid molecules dynamics depends strongly on temperature and cholesterol concentration. At 323K and short times, t∼0.1μs the lipid molecules’ feature an anomalous sub-diffusive behavior with the mean square displacement (MSD) scaling as ∼t1/2. The characteristic time needed to reach the diffusive regime MSD ∼t, increases with the cholesterol content, reaching 0.2μs for 40% cholesterol content. The lipid diffusion activation energy in the Lo phase is 57kJ/mol, in good agreement with the available experimental estimates.
Finite-size and truncation effects for microscopic expressions for the temperature at equilibrium and nonequilibrium
Anders Lervik, Øivind Wilhelmsen, Thuat T. Trinh and Henrik Rojas Nagel
J. Chem. Phys., 143:114106, 2015 | Link to article
View summary
Several expressions have been proposed for the temperature in molecular simulations, where some of them have configurational contributions. We investigate how their accuracy is influenced by the number of particles in the simulation and the discontinuity in the derivatives of the interaction potential introduced by truncation. For equilibrium molecular dynamics with fixed total volume and fixed average total energy per particle, all the evaluated expressions including that for the kinetic temperature give a dependence on the total number of particles in the simulation. However, in a partitioned simulation volume under the same conditions, the mean temperature of each bin is independent of the number of bins. This finding is important for consistently defining a local temperature for use in nonequilibrium simulations. We identify the configurational temperature expressions which agree most with the kinetic temperature and find that they give close to identical results in nonequilibrium molecular dynamics (NEMD) simulations with a temperature gradient, for high and low density bulk-systems (both for transient and steady-state conditions), and across vapor-liquid interfaces, both at equilibrium and during NEMD simulations. The work shows that the configurational temperature is equivalent to the kinetic temperature in steady-state molecular dynamics simulations if the discontinuity in the derivatives of the interaction potential is handled properly, by using a sufficiently long truncation-distance or tail-corrections.
Michaelis-Menten kinetics under non-isothermal conditions
Anders Lervik, Signe Kjelstrup and Hong Qian
Phys. Chem. Chem. Phys., 17:1317-1324, 2015 | Link to article
View summary
We extend the celebrated Michaelis-Menten kinetics description of an enzymatic reaction taking into consideration the presence of a thermal driving force. A coupling of chemical and thermal driving forces is expected from the principle of non-equilibrium thermodynamics, and specifically we obtain an additional term to the classical Michaelis-Menten kinetic equation, which describes the coupling in terms of a single parameter. A companion equation for the heat flux is also derived, which actually can exist even in the absence of a temperature difference. Being thermodynamic in nature, this result is general and independent of the detailed mechanism of the coupling. Conditions for the experimental verification of the new equation are discussed.
Gluing Potential Energy Surfaces with Rare Event Simulations
Anders Lervik and Titus S. van Erp
J. Chem. Theory Comput., 11:2440-2450, 2015 | Link to article
View summary
We develop a new method combining replica exchange transition interface sampling with two distinct potential energy surfaces. The method can be used to combine different levels of theory in a simulation of a molecular process (e.g., a chemical reaction), and it can serve as a dynamical version of QM-MM, connecting classical dynamics with Ab Initio dynamics in the time domain. This new method, which we coin QuanTIS, could be applied to use accurate but expensive density functional theory based molecular dynamics for the breaking and making of chemical bonds, while the diffusion of reactants in the solvent are treated with classical force fields. We exemplify the method by applying it to two simple model systems (an ion dissociation reaction and a classical hydrogen model), and we discuss a possible extension of the method in which classical force field parameters for chemical reactions can be optimized on the fly.
2014 (1)#
Sorting particles with nanoscale thermophoretic devices: how efficient is it?
Anders Lervik and Fernando Bresme
Phys. Chem. Chem. Phys., 16:13279-13286, 2014 | Link to article
View summary
We investigate particle separation driven by thermal gradients across solid state nanopores using a combined molecular dynamics simulation, non-equilibrium thermodynamics theory and a kinetic model approach. The thermophoretic device, a thermal nanopump, exploits thermal gradients to sort particles of different mass, which accumulate preferentially in hot or cold reservoirs. We show that the large amount of energy dissipated by the thermal nanopump during the transport process leads in general to very low efficiencies, 0.01-0.15%. We find that the nanopump thermal conductivity and structure plays a crucial role in determining the efficiency and a route to enhance it. Doubling the pore radius, from 0.5-1 nm radius, leads to a large increase in the mass diffusion and to a 20 fold increase in the efficiency. Addition of nanoscale defects, without modification of the nanopore structure, leads to a large reduction of the nanopump thermal conductivity and to a large enhancement of the thermodynamic efficiency. We find that nanopumps with nanoscale defects are >3 times more efficient than those without defects. Finally, we identify the microscopic variables responsible for the enhancement of thermally induced transport across nanopores and discuss strategies to tune these variable in order to regulate transport efficiency.
2013 (3)#
Describing transport across complex biological interfaces
A. Lervik and S. Kjelstrup
Eur. Phys. J. Special Topics, 222:143--159, 2013 | Link to article
View summary
It has long been known that proteins are capable of transporting ions against a gradient in the chemical potential, using the energy available from a chemical reaction. This is called active transport. A well studied example is the Ca2+-transport by means of hydrolysis of adenosine triphoshpate (ATP) at the surface of the Ca2+-ATPase in sarcoplasmic reticulum. The cycle of events is known to be reversible, and has recently also been associated with a characteristic, and also reversible, heat production. We use the case of the Ca2+-ATPase to present and discuss various central theoretical approaches to describe active transport, with focus on two schools of development, namely the kinetic and the thermodynamic schools. Among the kinetic descriptions, Hill's diagram method gives the most sophisticated description, reducing to the common Post-Albers scheme with simple enzyme kinetic reactions. Among the thermodynamic approaches, we review the now classical approach of Katchalsky and Curran, and its extension to proper pathways by Caplan and Essig, before the most recent development based on mesoscopic theory is outlined. The mesoscopic approach gives a non-linear theory compatible with Hill's most general method when the active transport is isothermal. We show how the old question of scalar-vector coupling is resolved using rules for non-equilibrium thermodynamics for interfaces. Also thermal driving forces can then be accounted for. Essential physical concepts behind all methods are presented and advantages/deficiencies are pointed out. Emphasis is made on the connection to experiments.
Active transport of the Ca2+-pump: introduction of the temperature difference as a driving force
Anders Lervik, Dick Bedeaux and Signe Kjelstrup
Eur. Biophys. J., 42:321--331, 2013 | Link to article
View summary
We analyse a kinetic cycle of the Ca2+-ATPase molecular pump using mesoscopic non-equilibrium thermodynamics. The pump is known to generate heat, and by analysing the operation on the mesoscopic level, we are able to introduce a temperature difference and the corresponding heat flux in the description. Integration over the internal coordinates then results in non-linear flux--force relations describing the operation of the pump on the macroscopic level. Specifically, we obtain an expression for the heat flux associated with the active transport and the coupling of heat effects to the transport of ions and the rate of the ATP-hydrolysis.
Enhancement of the Thermal Polarization of Water via Heat Flux and Dipole Moment Dynamic Correlations
Jeff Armstrong, Anders Lervik and Fernando Bresme
J. Phys. Chem. B, 117:14817-14826, 2013 | Link to article
View summary
It has been recently shown that liquid water polarizes as a response to a temperature gradient. This polarization effect can be significant for temperature gradients that can be achieved at micro and nanoscales. In this paper we investigate the dependence of the polarization response of liquid and supercritical water at different thermodynamic conditions using both equilibrium and nonequilibrium molecular dynamics simulations for the extended point charge water model. We find that the thermal polarization features a nonmonotonic behavior with temperature, reaching a maximum response at specific thermodynamic states. We show that the thermal polarization is maximized when the density of states of the heat flux and dipole moment correlation functions feature the strongest overlap. The librational modes of water are shown to play an important role in determining this behavior as well as the heat transport mechanism in water. The librational frequencies show a significant dependence with temperature and pressure. This dependence provides a microscopic mechanism to explain the observed maximization of the thermal-polarization effect. Our work provides new microscopic insights on the mechanism determining the orientation of polar fluids under thermal gradients, as well as new strategies to maximize their orientation by manipulating the dynamic correlations between the heat flux and the sample dipole moment.
2012 (4)#
Kinetic and mesoscopic non-equilibrium description of the Ca2+ pump: a comparison
Anders Lervik, Dick Bedeaux and Signe Kjelstrup
Eur. Biophys. J., 41:437--448, 2012 | Link to article
View summary
We analyse the operation of the Ca2+-ATPase ion pump using a kinetic cycle diagram. Using the methodology of Hill, we obtain the cycle fluxes, entropy production and efficiency of the pump. We compare these results with a mesoscopic non-equilibrium description of the pump and show that the kinetic and mesoscopic pictures are in accordance with each other. This gives further support to the mesoscopic theory, which is less restricted and also can include the heat flux as a variable. We also show how motors can be characterised in terms of unidirectional backward fluxes. We proceed to show how the mesoscopic approach can be used to identify fast and slow steps of the model in terms of activation energies, and how this can be used to simplify the kinetic diagram.
Molecular dynamics simulations of the Ca2+-pump: a structural analysis
Anders Lervik, Fernando Bresme and Signe Kjelstrup
Phys. Chem. Chem. Phys., 14:3543-3553, 2012 | Link to article
View summary
We report large scale molecular dynamics computer simulations, [similar]100 ns, of the ion pump Ca2+-ATPase immersed in a 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) bilayer. The structure simulated here, E1, one of the several conformations resolved using X-ray diffraction techniques, hosts two Ca2+-ions in the hydrophobic domain. Our results indicate that protonated residues lead to stronger ion-residue interactions, supporting previous conclusions regarding the sensitivity of the Ca2+ behaviour to the protonated state of the amino acid binding sites. We also investigate how the protein perturbs the bilayer structure. We show that the POPC bilayer is [similar]12% thinner than the pure bilayer, near the protein surface. This perturbation decays exponentially with the distance from the protein with a characteristic decay length of 0.8 nm. We find that the projected area per lipid also decreases near the protein. Using an analytical model we show that this change in the area is only apparent and it can be explained by considering the local curvature of the membrane. Our results indicate that the real area per lipid near the protein is not significantly modified with respect to the pure bilayer result. Further our results indicate that the local deformation of the membrane around the protein might be compatible with the enhanced protein activity observed in experiments over a narrow range of membrane thicknesses.
Nonequilibrium molecular dynamics simulations of the thermal conductivity of water: A systematic investigation of the SPC/E and TIP4P/2005 models
Frank Römer, Anders Lervik and Fernando Bresme
J. Chem. Phys., 137:074503, 2012 | Link to article
View summary
We report an extensive nonequilibrium molecular dynamics investigation of the thermal conductivity of water using two of the most accurate rigid nonpolarizable empirical models available, SPC/E and TIP4P/2005. Our study covers liquid and supercritical states. Both models predict the anomalous increase of the thermal conductivity with temperature and the thermal conductivity maximum, hence confirming their ability to reproduce the complex anomalous behaviour of water. The performance of the models strongly depends on the thermodynamic state investigated, and best agreement with experiment is obtained for states close to the liquid coexistence line and at high densities and temperatures. Considering the simplicity of these two models the overall agreement with experiments is remarkable. Our results show that explicit polarizability and molecular flexibility are not needed to reproduce the anomalous heat conduction of water.
On the Thermodynamic Efficiency of Ca2+-ATPase Molecular Machines
Anders Lervik, Fernando Bresme, Signe Kjelstrup and J. Miguel Rubí
Biophys. J., 103:1218 - 1226, 2012 | Link to article
View summary
Experimental studies have shown that the activity of the reconstituted molecular pump Ca2+-ATPase strongly depends on the thickness of the supporting bilayer. It is thus expected that the bilayer structure will have an impact on the thermodynamic efficiency of this nanomachine. Here, we introduce a nonequilibrium-thermodynamics theoretical approach to estimate the thermodynamic efficiency of the Ca2+-ATPase from analysis of available experimental data about ATP hydrolysis and Ca2+ transport. We find that the entropy production, i.e., the heat released to the surroundings under working conditions, is approximately constant for bilayers containing phospholipids with hydrocarbon chains of 18–22 carbon atoms. Our estimates for the heat released during the pump operation agree with results obtained from separate calorimetric experiments on the Ca2+-ATPase derived from sarcoplasmic reticulum. We show that the thermodynamic efficiency of the reconstituted Ca2+-ATPase reaches a maximum for bilayer thicknesses corresponding to maximum activity. Surprisingly, the estimated thermodynamic efficiency is very low, ∼12%. We discuss the significance of this result as representative of the efficiency of other nanomachines, and we address the influence of the experimental set-up on such a low efficiency. Overall, our approach provides a general route to estimate thermodynamic efficiencies and heat dissipation in experimental studies of nanomachines.
2010 (1)#
Heat transfer in protein-water interfaces
Anders Lervik, Fernando Bresme, Signe Kjelstrup, Dick Bedeaux and J. Miguel Rubi
Phys. Chem. Chem. Phys., 12:1610-1617, 2010 | Link to article
View summary
We investigate using transient non-equilibrum molecular dynamics simulation the temperature relaxation process of three structurally different proteins in water, namely; myoglobin, green fluorescence protein (GFP) and two conformations of the Ca2+-ATPase protein. By modeling the temperature relaxation process using the solution of the heat diffusion equation we compute the thermal conductivity and thermal diffusivity of the proteins, as well as the thermal conductance of the protein-water interface. Our results indicate that the temperature relaxation of the protein can be described using a macroscopic approach. The protein-water interface has a thermal conductance of the order of 100-270 MW K-1 m-2, characteristic of water-hydrophilic interfaces. The thermal conductivity of the proteins is of the order of 0.1-0.2 W K-1 m-1 as compared with [approximate]0.6 W K-1 m-1 for water, suggesting that these proteins can develop temperature gradients within the biomolecular structures that are larger than those of aqueous solutions. We find that the thermal diffusivity of the transmembrane protein, Ca2+-ATPase is about three times larger than that of myoglobin or GFP. Our simulation shows that the Kapitza length of these structurally different proteins is of the order of 1 nm, showing that the protein-water interface should play a major role in defining the thermal relaxation of biomolecules.
2009 (1)#
Heat transfer in soft nanoscale interfaces: the influence of interface curvature
Anders Lervik, Fernando Bresme and Signe Kjelstrup
Soft Matter, 5:2407-2414, 2009 | Link to article
View summary
We investigate, using transient non-equilibrium molecular-dynamics simulations, heat-transfer through nanometer-scale interfaces consisting of n-decane (2-12 nm diameter) droplets in water. Using computer simulation results of the temperature relaxation of the nanodroplet as a function of time we have computed the thermal conductivity and the interfacial conductance of the droplet and the droplet/water interface respectively. We find that the thermal conductivity of the n-decane droplets is insensitive to droplet size, whereas the interfacial conductance shows a strong dependence on the droplet radius. We rationalize this behavior in terms of a modification of the n-decane/water surface-tension with droplet curvature. This enhancement in interfacial conductance would contribute, in the case of a suspension, to an increase in the thermal conductivity with decreasing particle radius. This notion is consistent with recent experimental studies of nanofluids. We also investigate the accuracy of different diffusion equations to model the temperature relaxation in non stationary non equilibrium processes. We show that the modeling of heat transfer across a nanodroplet/fluid interface requires the consideration of the thermal conductivity of the nanodroplet as well as the temperature discontinuity across the interface. The relevance of this result in diffusion models that neglect thermal conductivity effects in the modeling of the temperature relaxation is discussed.
2008 (1)#
Water Polarization under Thermal Gradients
Fernando Bresme, Anders Lervik, Dick Bedeaux and Signe Kjelstrup
Phys. Rev. Lett., 101:020602, 2008 | Link to article
View summary
We investigate the response of bulk liquid water to a temperature gradient using nonequilibrium molecular dynamics simulations. It is shown that the thermal gradient polarizes water in the direction of the gradient, leading to a non-negligible electrostatic field whose origin lies in the water reorientation under nonequilibrium conditions. The dependence of the magnitude of the electrostatic field with the temperature gradient is in agreement with nonequilibrium thermodynamics theory. We conclude that temperature gradients of the order of 10^8 K/m could result in fairly large polarizations ~ 10^6 V/m.